变分量子特征求解算法(VQE)
变分量子特征值求解算法(Variational-Quantum-Eigensolver,简称VQE)[1, 2]是用于找到一个矩阵 \(H\) (通常是一个较大矩阵)的特征值的量子与经典混合的算法。 当这个算法用在量子模拟时,\(H\) 就是某系统的Hamiltonian [3, 4, 5]。在这个混合算法中,量子子程序是在经典优化回路内部运行。
这里的量子子程序有两个基本的步骤:
(1)制备量子态 \(|\Psi(\vec{\theta})\rangle\) 。
(2)测量期望值 \(\langle\,\Psi(\vec{\theta})\,|\,H\,|\,\Psi(\vec{\theta})\,\rangle\) 。
由 变分原则 可知,这个期望值不小于 \(H\) 的最小特征值。 这个限定使得我们能够运用优化回路(optimization loop)的经典计算方法来找特征值:
(1)运用经典的非线性优化器通过改变参数 来最小化期望值 \(\vec{\theta}\) 。
(2)不断迭代,直到收敛。
事实上,VQE的量子子程序等价于基于参数 \(\vec{\theta}\) 的集合来制备一个状态,并在适当的基上进行一系列的测量。在这个算法中参数化的状态的制备是比较困难的,并且参数化状态的制备显著地影响到算法的工作性能。
接口介绍
在QPanda中我们实现了上述算法,使用该算法必须包含QPanda命名空间 QPanda::VQE,详见 VQE.h 。
-
class VQE
-
VQE(OptimizerType optimizer = OptimizerType::Powell)
- 功能
构造函数。通过传入指定的优化器类型来进行构造,默认使用的优化器是
Powell- 参数
optimizer 优化器类型
-
VQE(const std::string &optimizer)
- 功能
构造函数。通过传入string类型的优化器来进行构造。
- 参数
optimizer 优化器类型
-
void setMoleculeGeometry(const QMoleculeGeometry &geometry)
- 功能
设置分子结构,将分子结构转换成
QMoleculeGeometry的形式进行输入。- 参数
geometry 分子结构
- 返回值
无
-
void setAtomsPosGroup(const QAtomsPosGroup &pos)
- 功能
设置该分子的一组原子坐标,算法将会计算每个原子坐标对应的能量。
- 参数
pos 一组原子坐标
- 返回值
无
-
void setBasis(const std::string &basis)
- 功能
设置psi4应用全局配置选项basis。
- 参数
basis psi4配置
- 返回值
无
-
void setMultiplicity(const int &multiplicity)
- 功能
设置分子重数 multiplicity (defined as 2S + 1)。
- 参数
multiplicity 分子重数
- 返回值
无
-
void setCharge(const int &charge)
- 功能
设置分子的电子数。
- 参数
charge 电子数
- 返回值
无
-
void setPsi4Path(const std::string &path)
- 功能
设置Psi4应用的路径。
- 参数
path Psi4应用的路径
- 返回值
无
-
void setDataSavePath(const std::string &path)
- 功能
设置计算结果保存路径,每个分子结构计算的结果将以result_[index].dat的命名方式进行保存,其中"index"表示分子结构的索引号。
- 参数
path 计算结果保存路径
- 返回值
无
-
void enableOptimizerData(bool enable)
- 功能
是否保存中间优化计算结果,存放的路径为setDataSavePath配置的路径。
- 参数
enable 若配置为true则保存,否则不保存
- 返回值
无
-
bool exec()
- 功能
执行算法。
- 参数
无
- 返回值
返回true表示VQE成功执行,否则执行失败。
-
vector_d getEnergies() const
- 功能
获取通过setAtomsPosGroup配置的一组坐标对应的能量值。
- 参数
无
- 返回值
一组能量值。
-
std::string getLastError()
- 功能
获取最后一次错误信息。
- 参数
无
- 返回值
最后一次错误信息。
-
AbstractOptimizer *getOptimizer()
- 功能
获取优化器实例,通过该实例修改优化器的参数。
- 参数
无
- 返回值
优化器实例指针。
-
VQE(OptimizerType optimizer = OptimizerType::Powell)
实例
下面我们通过计算一组氢分子的坐标,来展示如何使用VQE算法的接口。
备注
运行VQE算法必须要在系统上配置好psi4应用程序和python环境。
针对2原子分子获得一组原子坐标
QPanda::QAtomsPosGroup get2AtomPosGroup(double begin, double end, int size)
{
QPanda::QAtomsPosGroup atoms_pos_group;
QPanda::QPosition first(0, 0, 0);
double delta = (end - begin) / size;
for (int i = 0; i < size; i++)
{
std::vector<QPanda::QPosition> vec;
QPanda::QPosition second(0, 0, 0);
second.z = begin + i * delta;
vec.push_back(first);
vec.push_back(second);
atoms_pos_group.push_back(vec);
}
return atoms_pos_group;
}
调用VQE算法接口,求解给定一组坐标的能量
#include "VQE/VQE.h"
#include "Optimizer/AbstractOptimizer.h"
int main()
{
const double begin = 0.25;
const double end = 2.5;
const size_t cnt = 50;
auto pos_group = get2AtomPosGroup(begin, end, cnt);
QPanda::QMoleculeGeometry geometry
{
{"H",{0, 0, 0}},
{"H",{0, 0, 0.74}}
};
std::string psi4_path = "D:/psi4/bin/psi4";
QPanda::VQE vqe;
vqe.setMoleculeGeometry(geometry);
vqe.setAtomsPosGroup(pos_group);
vqe.setPsi4Path(psi4_path);
vqe.setMultiplicity(1);
vqe.setCharge(0);
vqe.setBasis("sto-3g");
vqe.getOptimizer()->setDisp(true);
vqe.exec();
auto energies = vqe.getEnergies();
if (pos_group.size() != energies.size())
{
std::cout << "VQE failed! Last error: " << vqe.getLastError() << std::endl;
}
else
{
for (auto i = 0u; i < energies.size(); i++)
{
std::cout << energies[i] << std::endl;
}
}
return 0;
}
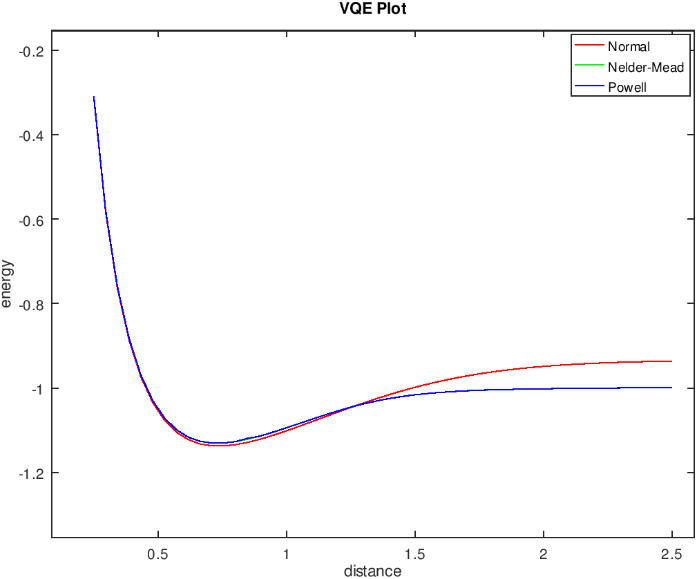
我们也可以配置 Nelder-Mead 优化器进行优化,下图是上述分子结构使用不同的优化器的效果